
티스토리 뷰
1. SDM 생물종 분포모델 개발하기
6) 생물종 분포모델 실행하기
# SDM 패키지 불러오기
library(sdm)
# SMD 패키지에 적합한 입력데이터와 환경변수 만들기
d <- sdmData(sp~., sp, predictors= bioc, bg = list(method='gRandom',n=1000))
d- sdm 데이터 정보 요약 : 생물종 1종, 환경변수 19개, 환경변수 이름, 종분포모델 유형: 출현과 배경데이터 등

SMD 패키지에서 사용가능한 모델 방법을 확인하기 위해서 다음 명령어 구문 실행
getmethodNames()
7) SDM 생물종 모델 개발하기
- 이 글에서 사용한 모델 방법은 'glm','brt','rf','tree','mars','maxent','svm' 등 7가지를 이용하고
검증자료는 30%로 서브샘플링과 부트스트랩 검증방법을 이용한다.
다음 구문은 7개 모델이 2가지 검증방법을 3번 반복함으로써 각 모델방법에 따라 6가지 결과를 가져온다.
m <- sdm(sp~., d, methods=c('glm','brt','rf','tree','mars','maxent','svm'), replication=c('sub','boot'), test.p=30,n=3, parallelSetting=list(ncore=4,method='parallel'))
m
모델 개발결과

8) 모델 방법의 정확도를 그래프로 확인하기 위해서 다음 구문을 실행한다.
roc(m[[c(4,10,16,22,28,34,40)]])

7) 현재 기후환경 데이터 기반 생물종 분포모델 실행하여 예측하기
- 7개 모델에 따라 생성된 예측결과를 평균하여 예측하기
p2 <- predict(m, bio,filename='pr_Brachydiplax.img' ,mean=T ,overwrite=TRUE)
p2 names(p2)
plot(p2)
- 미래 분포지역과 비교하기 위해서 7개 모델의 장단점을 이용한 앙상블 모델로 분포예측
en <- ensemble(m, p, filename='en_Brachydiplax_bioc.img',setting=list(method='weighted',stat='tss',opt=2))
cl <- colorRampPalette(c('#3E49BB','#3498DB','yellow','orange','red','darkred'))
plot(en , col=cl(200))
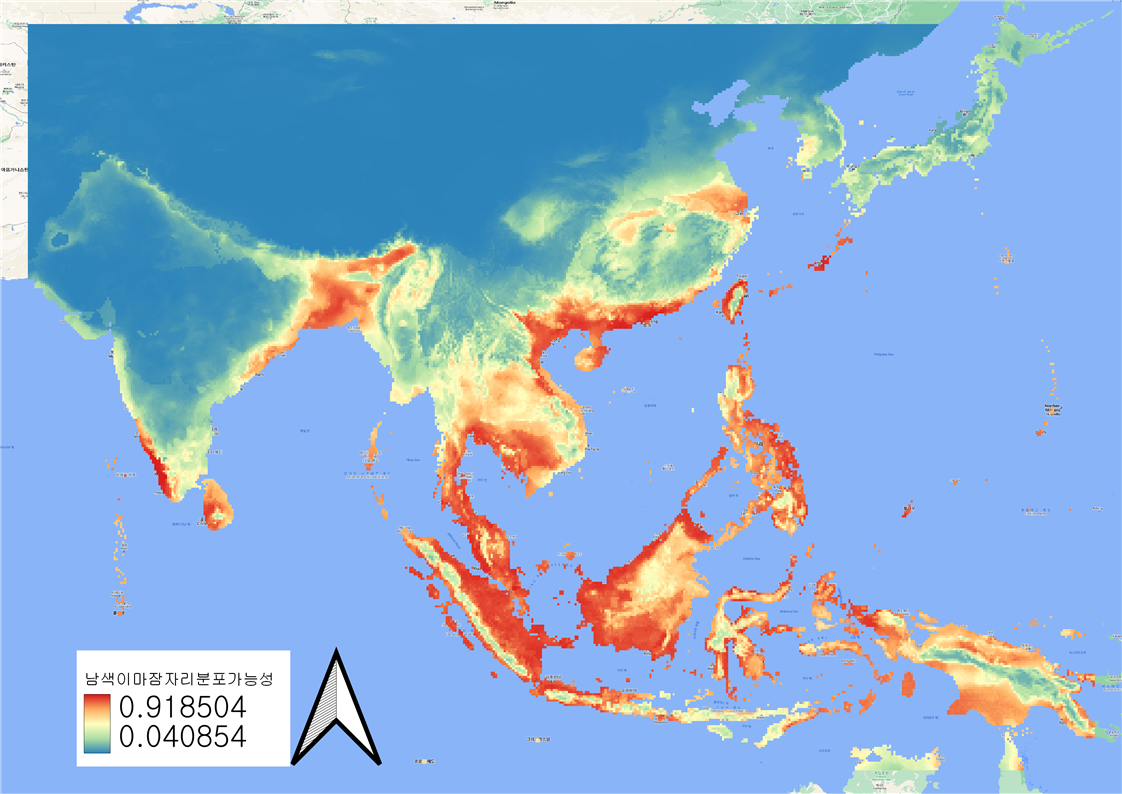
7) 미래 기후환경 데이터 기반 생물종 분포모델 실행하기
# 미래기후자료 불러와서 대상지역으로 한정하기
biof <- raster::getData('CMIP5', var='bio', res=10, rcp=85, model='CN', year=70)
biof
plot(biof[[1]])
names(biof) <- names(bio)
# 대상지역으로 한정하기
biofc <- crop(biof, e)
- 미래 분포지역을 예측하기 위해 7개 모델의 장단점을 이용한 앙상블 모델로 분포예측
en1 <- ensemble(m, biofc, filename='enf_future.img',setting=list(method='weighted',stat='tss',opt=2))
plot(en1, col=cl(200))

8) 현재와 미래의 분포가능성 비교 분석하기
ch <- en1 - en
cl <- colorRampPalette(c('red','orange','yellow','gray','green','blue'))
plot(ch,col=cl(200))

2. SDM 전체 실행 코드
# 남색이마잠자리 분포변화 예측
# Error in .updateGDAL(object, v, cell, band, setminmax) :
# no longer supported
# devtools::install_url('https://cran.r-project.org/src/contrib/Archive/raster/raster_3.5-15.tar.gz')
# devtools::install_url('https://cran.r-project.org/src/contrib/Archive/dismo/dismo_1.3-5.tar.gz')
# sdm 패키지 설치
# install.packages('sdm')
# or
# devtools::install_github('babaknaimi/sdm')
# 라이브러리 불러오기
library(sdm)
#installAll() # only firest time after installing the sdm package
library(dismo)
library(dplyr)
library(tidyr)
library(mapview)
library(raster)
library(rgeos)
# 작업폴더 설정
setwd("c:/R_work")
getwd()
#남색이마잠자리 shp 파일 지정
file <- "./남색이마잠자리_pt.shp"
# shape 파일 불러오기
sp <- shapefile(file)
head(sp)
class(sp)
# bioclim data 해상도 10분(해상도 300km) 짜리 다운로드하기
bio <- raster::getData('worldclim', var='bio', res=10)
bio
names(bio)
# bio 변수 1번인 평균기온 출력하고 그 위에 남색이마잠자리 위치포인터 올리기
plot(bio[[1]])
points(sp)
class(bio[[1]])
# 대상지역 한정하기
ext <- "./남색이마잠자리_AOI.shp" # shape 파일 지정
sp_ext <- shapefile(ext) # shape 파일 불러오기
head(sp_ext)
class(sp_ext)
# 직접 좌표값을 xmin, xmax, ymin, ymax 순으로 입력해서 사각형 영역 설정
# e <- extent(-25,60,-50,50)
# e
e <- extent(sp_ext)
e
# 마우스를 이용해서 사각형 영역 설정
# e <- drawExtent()
spg <- crop(sp, e)
points(sp,col='red')
bioc <- crop(bio, e)
plot(bioc[[1]])
points(sp)
# 기후자료 GIS 래스터 asc파일로 저장하기
for (x in 1:19) {
print(names(bioc[[x]]))
writeRaster(bioc[[x]], paste('wc10_asc/','bio_',x,'.asc'), NAflag=-9999, overwrite=TRUE)
}
#환경변수간 다중공선성 체크하기
library(usdm)
vif(bioc)
ex <- raster::extract(bioc,spg)
class(ex)
head(ex)
head(spg)
ex_df = as.data.frame(ex)
ex_df
coordinates(ex_df) <- c('lon','lat')
raster::shapefile(ex_df, "ex_val.shp")
v <- vifstep(ex)
#vifcor
v
bioc <- exclude(bioc, v)
#생물종 분포모델 개발 시작-
library(sdm)
head(sp)
d <- sdmData(sp~., sp, predictors= bioc, bg = list(method='gRandom',n=1000))
d
getmethodNames()
m <- sdm(sp~., d, methods=c('glm','brt','rf','tree','mars','maxent','svm'), replication=c('sub','boot'),
test.p=30,n=3, parallelSetting=list(ncore=4,method='parallel'))
m
roc(m[[c(1,7,13,19,25,31,37)]])
roc(m[[c(4,10,16,22,28,34,40)]])
#m@models$species$rf$`13`@object
gui(m)
p <- predict(m, bioc,filename='pr_Brachydiplax_bioc.img' ,overwrite=TRUE)
p
names(p)
plot(p[[c(1,7,13,19,25,31,37)]])
plot(p[[c(4,10,16,22,28,34,40)]])
p1 <- predict(m, bio,filename='pr_Brachydiplax.img' ,overwrite=TRUE)
p1
names(p1)
plot(p1[[c(1,7,13,19,25,31,37)]])
p2 <- predict(m, bio,filename='pr_Brachydiplax.img' ,mean=T ,overwrite=TRUE)
p2
names(p2)
plot(p2)
#en1 <- ensemble(m, bio, filename='en.img',setting=list(method='weighted',stat='tss',opt=2))
en <- ensemble(m, p, filename='en_Brachydiplax_bioc.img',setting=list(method='weighted',stat='tss',opt=2))
cl <- colorRampPalette(c('#3E49BB','#3498DB','yellow','orange','red','darkred'))
plot(en , col=cl(200))
# 미래기후자료 불러와서 대상지역으로 한정하기
biof <- raster::getData('CMIP5', var='bio', res=10, rcp=85, model='CN', year=70)
biof
plot(biof[[1]])
names(biof) <- names(bio)
biofc <- crop(biof, e)
for (x in 1:19) {
print(names(biofc[[x]]))
writeRaster(biofc[[x]], paste('future_CMIP5_asc/','bio_',x,'.asc'), NAflag=-9999, overwrite=TRUE)
}
en1 <- ensemble(m, biofc, filename='enf_future.img',setting=list(method='weighted',stat='tss',opt=2))
#--------------
plot(en1, col=cl(200))
cl <- colorRampPalette(c('#3E49BB','#3498DB','yellow','orange','red','darkred'))
#------
plot(en, col=cl(200))
plot(en1, col=cl(200))
proj4string(spg) <- projection(en)
library(mapview)
mapview(en,col.regions=cl(200)) + spg
#-----------
ch <- en1 - en
cl <- colorRampPalette(c('red','orange','yellow','gray','green','blue'))
plot(ch,col=cl(200))
writeRaster(ch, filename = "chagne_results.tif", format="GTiff", overwrite=T)
'머신러닝 & 딥러닝' 카테고리의 다른 글
| R 프로그램을 이용해서 다중공선성 확인하기 - 2편 (0) | 2023.03.16 |
|---|---|
| R기반 래스터 다중공선성 확인하기 (0) | 2023.03.15 |
| R기반 SDM패키지를 이용한 생물종 분포모델 - 2편 (0) | 2023.03.05 |
| R기반 SDM 패키지를 이용한 생물종 분포 모델-1편 (0) | 2023.03.04 |
| 위성사진(Satellite Imagery) 자료 다루기 1편 (0) | 2023.02.24 |