
티스토리 뷰
SDM은 species distribution model의 약자이고, 종 분포 모델링을 위한 객체 지향의 재현 가능하고 확장 가능한 R 플랫폼입니다. 생물종 분포모델은 생물종이 서식하는 위치와 이와 해당하는 주변 환경요소간의 관계를 지리적 공간에 예측하는 기법입니다. 생물종 분포 예측모델은 반응하는 변수에 따라 회귀(regresstion)모델이 될수도 있고 분류(classification)가 될 수 도 있습니다.
이글에서는 R 통계 프로그램의 패키지 중 하나인 SDM 기능을 이용해서 생물종 분포 모델을 설명하고자 합니다.
그럼 제일 먼저 생물종을 선택하고 이 생물종이 나타난 지점의 위치를 수집하는 것으로 부터 생물종 분포모델을 시작한다.
그래서 이글에서는 잠자리를 대상으로 현재 기후조건과 미래 기후조건을 이용해서 생물종 분포변화를 예측하고자 한다.
1. 잠자리 위치데이터 수집
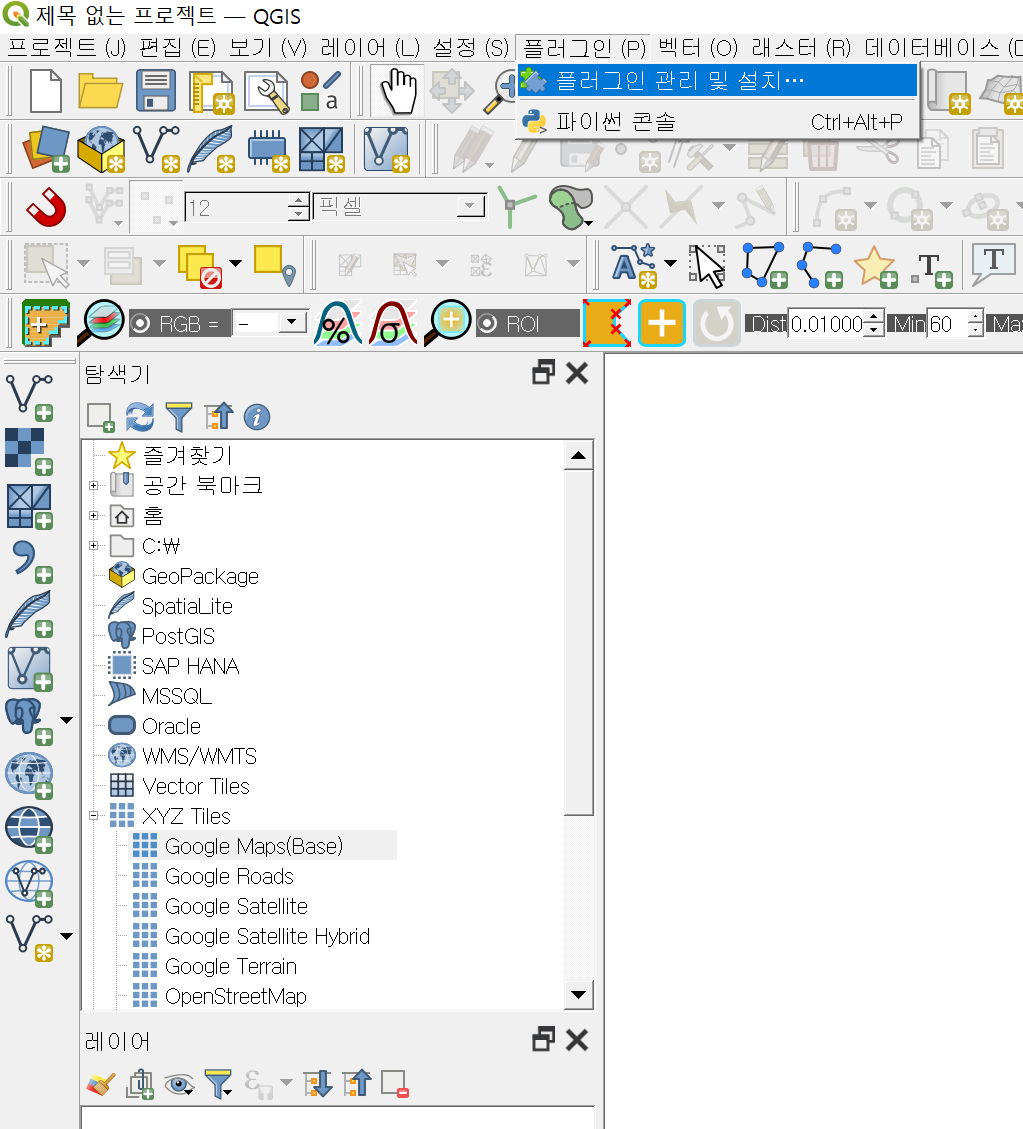
- 생물종 위치데이터는 QGIS의 플러그인 GBIF occurrence를 이용해서 수집한다.
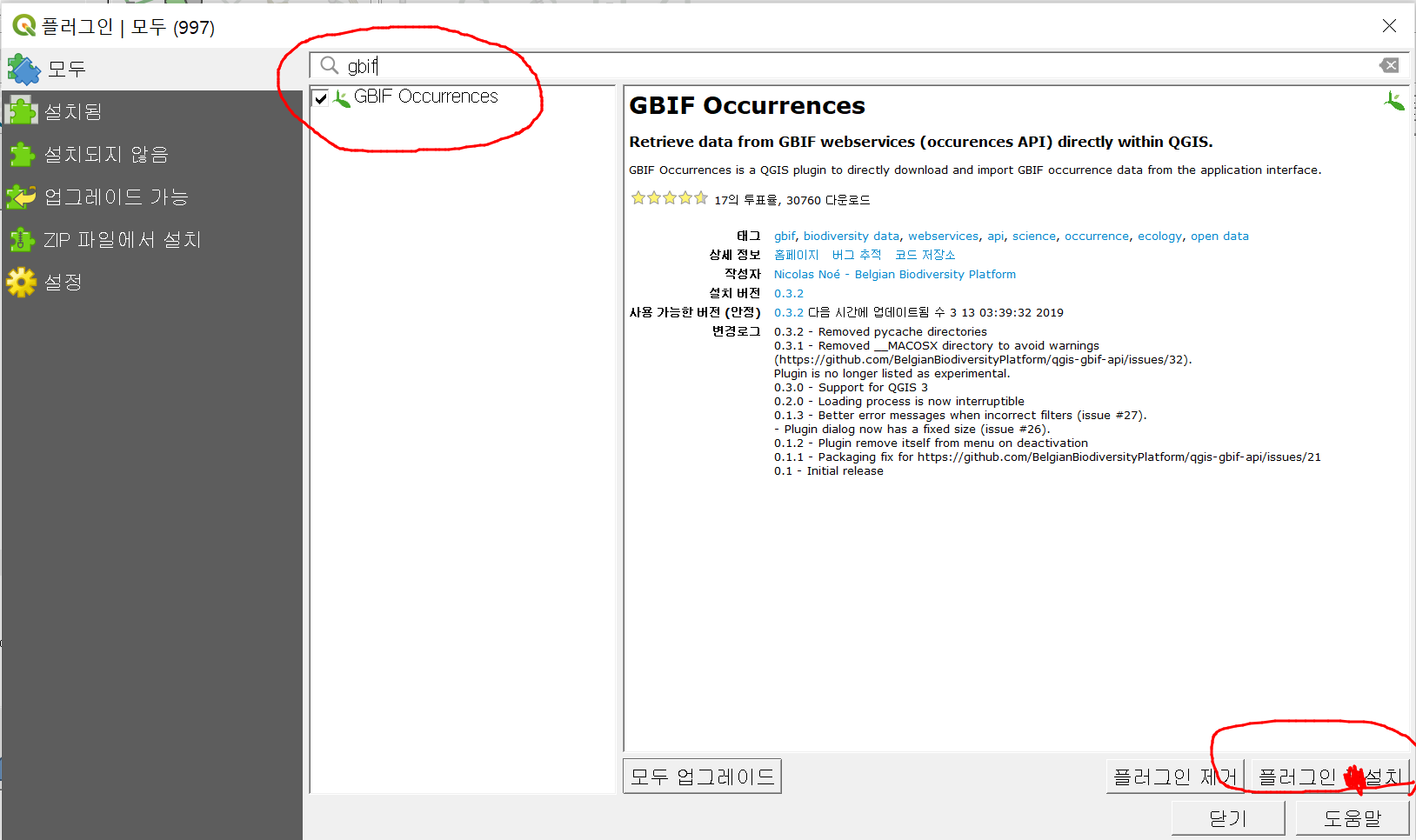
이를 위해서 QGIS프로그램에서 플러그인 설치를 이용하여 GBIF occurrence를 설치한다.
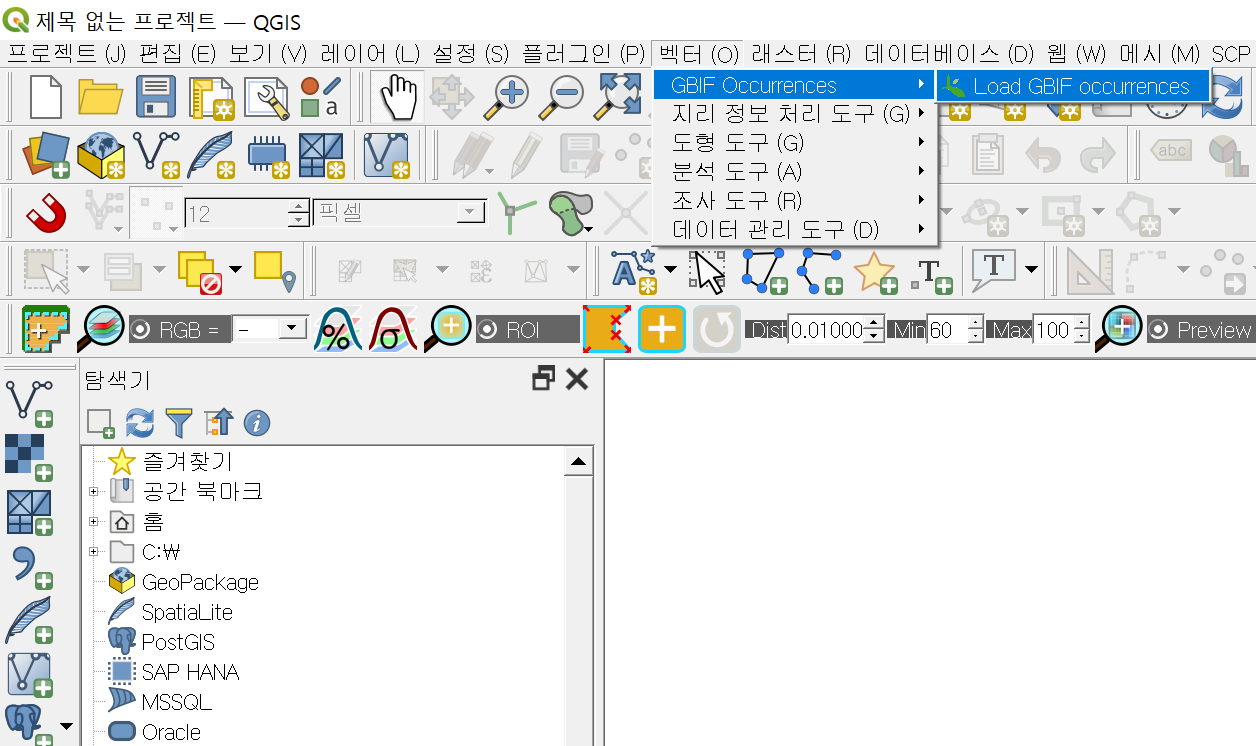
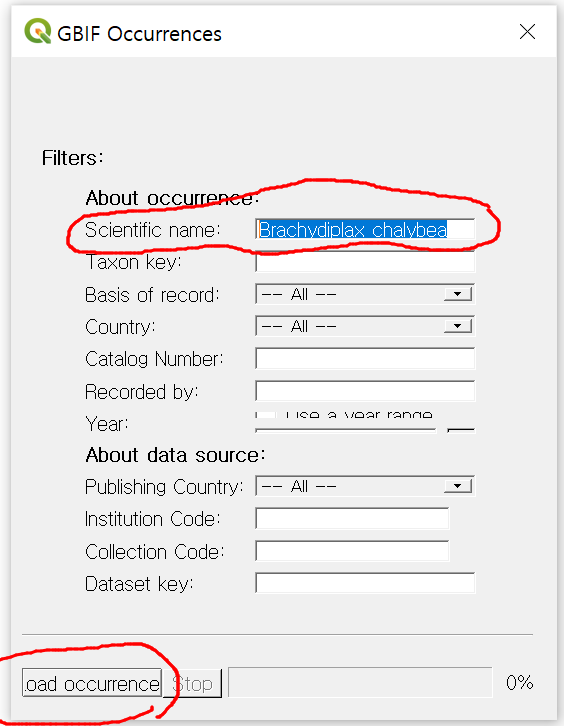
- 설치된 GBIF occurrecne 플러그인을 벡터메뉴에서 실행하고 나타난 대화상자에서 Scientific name 입력란에 잠자리에 대한 학명을 입력하고 load occurrence 버튼을 클릭한다.
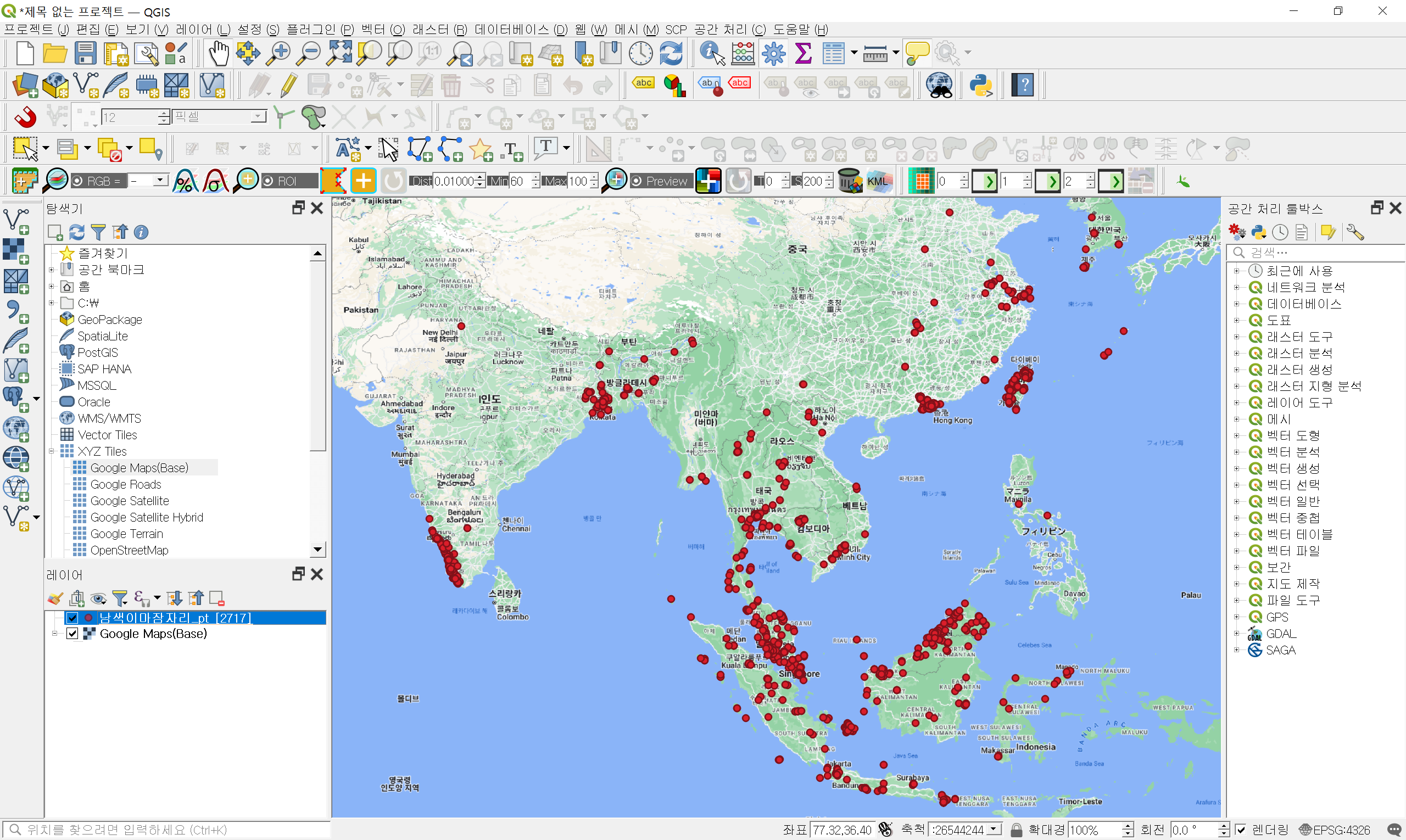
load occurrence 버튼을 클릭한 후, 다음 그림처럼 잠자리 위치자료가 검색되고 다운로드 된 다음 GIS shp파일로 변환되어 화면에 나타난다. 총 2717개의 잠자리 위치가 검색되고 다운되었다.
2. R 기반의 SDM 패키지 이용하여 생물종 분포모델 실행하기위한 환경설정
- 이글에서 사용된 R 프로그램 버전은 4.2.2이고 Rstudio 버전은 1.3.8이다.
- 현재 SDM을 사용하는데 다음 같은 오류가 발생한다.
- 더 자세한 방법은 이전 글 (2023.04.08 - [오류해결] - R 환경에서 생물종 분포모델 SDM에서 .updateGDAL 오류가 날 때 ) 을 참조하라.
# Error in .updateGDAL(object, v, cell, band, setminmax) : # no longer supported #
- 이 오류를 해결하기 위해서는 SDM에 필수적으로 연관된 R 패캐지인 raster와 disimo 패키지의 버전을 각각 3.5-15와 1.3-5버전으로 맞춰야 한다.
- SDM을 오류없이 실행하기 위해서는 raster와 disimo 패키지의 버전은 다음 명령어 구문으로 맞추면 된다.
- 만약 다음 구문에 오류가 있어 실행되지 않으면 rtools42-5355-5357.exe 파일을 다운받아 먼저 설치하고 실행한다.
devtools::install_url('https://cran.r-project.org/src/contrib/Archive/raster/raster_3.5-15.tar.gz') devtools::install_url('https://cran.r-project.org/src/contrib/Archive/dismo/dismo_1.3-5.tar.gz')
devtools::install_url('https://cran.r-project.org/src/contrib/Archive/raster/raster_3.5-15.tar.gz')
devtools::install_url('https://cran.r-project.org/src/contrib/Archive/dismo/dismo_1.3-5.tar.gz')3. SDM 패키지 설치 및 입력데이터와 환경데이터 입력하기
2편으로 >>> 2023.03.05 - [머신러닝 & 딥러닝] - R기반 SDM패키지를 이용한 생물종 분포모델 - 2편
1) SDM 패키지에서 받아들이는 입력파일 형태로 GIS shp파일을 변경한다.
2) SDM 패키지를 R프로그램에서 설치한다.
3)SDM 패키지를 원할하게 실행하기 위해 부수적인 패키지를 InstallAll()함수로 설치한다.
4) 생물종 위치자료 불러오기
5) 현재 기후환경 데이터 불러오기
6) 생물종 분포모델 실행하기
7) 미래 기후환경 데이터 기반 생물종 분포모델 실행하기
8) 현재와 미래의 분포가능성 비교 분석하기
4. SDM 전체 실행 코드
# 남색이마잠자리 분포변화 예측
# Error in .updateGDAL(object, v, cell, band, setminmax) :
# no longer supported
# devtools::install_url('https://cran.r-project.org/src/contrib/Archive/raster/raster_3.5-15.tar.gz')
# devtools::install_url('https://cran.r-project.org/src/contrib/Archive/dismo/dismo_1.3-5.tar.gz')
# sdm 패키지 설치
# install.packages('sdm')
# or
# devtools::install_github('babaknaimi/sdm')
# 라이브러리 불러오기
library(sdm)
#installAll() # only firest time after installing the sdm package
library(dismo)
library(dplyr)
library(tidyr)
library(mapview)
library(raster)
library(rgeos)
# 작업폴더 설정
setwd("c:/R_work")
getwd()
#남색이마잠자리 shp 파일 지정
file <- "./남색이마잠자리_pt.shp"
# shape 파일 불러오기
sp <- shapefile(file)
head(sp)
class(sp)
# bioclim data 해상도 10분(해상도 300km) 짜리 다운로드하기
bio <- raster::getData('worldclim', var='bio', res=10)
bio
names(bio)
# bio 변수 1번인 평균기온 출력하고 그 위에 남색이마잠자리 위치포인터 올리기
plot(bio[[1]])
points(sp)
class(bio[[1]])
ext <- "./남색이마잠자리_AOI.shp" # shape 파일 지정
sp_ext <- shapefile(ext) # shape 파일 불러오기
head(sp_ext)
class(sp_ext)
# 직접 좌표값을 xmin, xmax, ymin, ymax 순으로 입력해서 사각형 영역 설정
# e <- extent(-25,60,-50,50)
# e
e <- extent(sp_ext)
e
# 마우스를 이용해서 사각형 영역 설정
# e <- drawExtent()
spg <- crop(sp, e)
points(sp,col='red')
bioc <- crop(bio, e)
plot(bioc[[1]])
points(sp)
for (x in 1:19) {
print(names(bioc[[x]]))
writeRaster(bioc[[x]], paste('wc10_asc/','bio_',x,'.asc'), NAflag=-9999, overwrite=TRUE)
}
#------------
library(usdm)
vif(bioc)
ex <- raster::extract(bioc,spg)
class(ex)
head(ex)
head(spg)
ex_df = as.data.frame(ex)
ex_df
coordinates(ex_df) <- c('lon','lat')
raster::shapefile(ex_df, "ex_val.shp")
v <- vifstep(ex)
#vifcor
v
bioc <- exclude(bioc, v)
#--------------------
library(sdm)
head(sp)
d <- sdmData(sp~., sp, predictors= bioc, bg = list(method='gRandom',n=1000))
d
getmethodNames()
m <- sdm(sp~., d, methods=c('glm','brt','rf','tree','mars','maxent','svm'), replication=c('sub','boot'),
test.p=30,n=3, parallelSetting=list(ncore=4,method='parallel'))
m
roc(m[[c(1,7,13,19,25,31,37)]])
roc(m[[c(4,10,16,22,28,34,40)]])
#m@models$species$rf$`13`@object
gui(m)
p <- predict(m, bioc,filename='pr_Brachydiplax_bioc.img' ,overwrite=TRUE)
p
names(p)
plot(p[[c(1,7,13,19,25,31,37)]])
plot(p[[c(4,10,16,22,28,34,40)]])
p1 <- predict(m, bio,filename='pr_Brachydiplax.img' ,overwrite=TRUE)
p1
names(p1)
plot(p1[[c(1,7,13,19,25,31,37)]])
p2 <- predict(m, bio,filename='pr_Brachydiplax.img' ,mean=T ,overwrite=TRUE)
p2
names(p2)
plot(p2)
#en1 <- ensemble(m, bio, filename='en.img',setting=list(method='weighted',stat='tss',opt=2))
en <- ensemble(m, p, filename='en_Brachydiplax_bioc.img',setting=list(method='weighted',stat='tss',opt=2))
cl <- colorRampPalette(c('#3E49BB','#3498DB','yellow','orange','red','darkred'))
plot(en , col=cl(200))
##################
biof <- raster::getData('CMIP5', var='bio', res=10, rcp=85, model='CN', year=70)
biof
plot(biof[[1]])
names(biof) <- names(bio)
biofc <- crop(biof, e)
for (x in 1:19) {
print(names(biofc[[x]]))
writeRaster(biofc[[x]], paste('future_CMIP5_asc/','bio_',x,'.asc'), NAflag=-9999, overwrite=TRUE)
}
en1 <- ensemble(m, biofc, filename='enf_future.img',setting=list(method='weighted',stat='tss',opt=2))
#--------------
plot(en1, col=cl(200))
cl <- colorRampPalette(c('#3E49BB','#3498DB','yellow','orange','red','darkred'))
#------
plot(en, col=cl(200))
plot(en1, col=cl(200))
proj4string(spg) <- projection(en)
library(mapview)
mapview(en,col.regions=cl(200)) + spg
#-----------
ch <- en1 - en
cl <- colorRampPalette(c('red','orange','yellow','gray','green','blue'))
plot(ch,col=cl(200))
writeRaster(ch, filename = "chagne_results.tif", format="GTiff", overwrite=T)
'머신러닝 & 딥러닝' 카테고리의 다른 글
| R기반 SDM패키지를 이용하여 생물종 분포모델 - 3편 (0) | 2023.03.07 |
|---|---|
| R기반 SDM패키지를 이용한 생물종 분포모델 - 2편 (0) | 2023.03.05 |
| 위성사진(Satellite Imagery) 자료 다루기 1편 (0) | 2023.02.24 |
| JSON 데이터와 CSV데이터를 랜덤으로 생성하기 (0) | 2023.02.12 |
| 자바(Java SE(JRE/JDK)) 무료 버전 확인 및 설치 (0) | 2023.01.18 |